機械学習によるヨウ素の結合エネルギーの予測
従来法の1.3億倍のスピードでの予測に成功
千葉大学大学院薬学研究院 中島誠也 助教及び根本哲宏 教授は、超原子価ヨウ素(注1)と呼ばれるヨウ素含有分子の結合エネルギーを人工知能により算出する予測モデルの構築に成功しました。
分子は原子と原子が結合して成り立っています。その結合の強さを表す値、すなわち結合エネルギーの算出は、分子の安定性や反応性の指標となる重要なパラメーターです。この方法によって、従来の計算方法(DFT計算(注2))に比べ、1.3億倍ものスピードで結合エネルギーを算出することが可能となりました。
この研究成果は、2021年10月12日にネイチャー・リサーチ社のオープンアクセス学術誌である「Scientific Reports」に掲載されました。
分子は原子と原子が結合して成り立っています。その結合の強さを表す値、すなわち結合エネルギーの算出は、分子の安定性や反応性の指標となる重要なパラメーターです。この方法によって、従来の計算方法(DFT計算(注2))に比べ、1.3億倍ものスピードで結合エネルギーを算出することが可能となりました。
この研究成果は、2021年10月12日にネイチャー・リサーチ社のオープンアクセス学術誌である「Scientific Reports」に掲載されました。
- 研究の背景
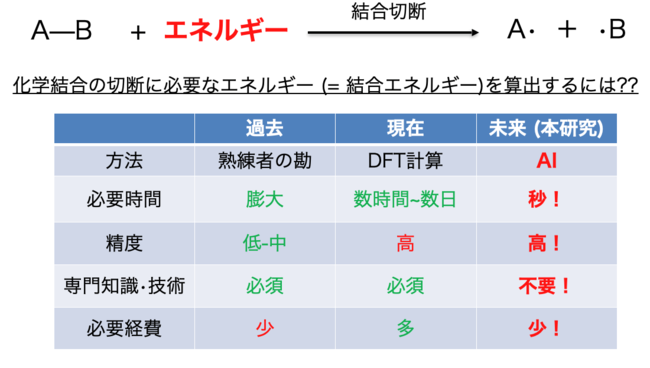 図1 結合エネルギー算出方法
図1 結合エネルギー算出方法
結合の強さは、その結合の切断に必要なエネルギーの値である「結合エネルギー」として数値で表すことができます。古くは実際の分子を用いて実測されてきましたが、近年はコンピュータの発展により、量子化学計算(主にDFT計算)によってシミュレーションすることが可能となりました (図1)。DFT計算では分子を実際に用意する必要がないため、架空の分子などの結合エネルギーも算出することが可能です。しかし、DFT計算では、3次元的な分子の最安定構造をコンピュータ上で算出し、その構造をもとに結合エネルギーを導くため、1つの分子の結合エネルギーの算出には数時間〜数日の計算時間が必要であり、分子の大きさが大きくなるほど、指数関数的に所要時間も膨れ上がります。そのため、DFT計算による結合エネルギーの算出には、高性能なコンピュータや高価なソフトウェア、そして計算に関する専門知識やノウハウが必須です。
そこで、誰でも簡単に結合エネルギーを算出できる方法論を開発することを目標とし、人工知能(AI)を用いる算出方法を着想しました。もし3次元的な構造を必要とせず、分子の構造を表す名前のような文字列から結合エネルギーが算出できれば、上記のすべての問題点が解決できると期待できます。
- 研究成果1- 超原子価ヨウ素の結合エネルギー予測モデルの構築
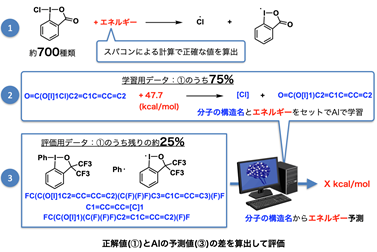 図2 予測モデルの構築方法
図2 予測モデルの構築方法
次にその75%を、分子の構造名と結合エネルギーの値をセットで機械学習を行い、結合エネルギー予測モデルを構築しました(図2②)。
その後、残りの25%を用いて、実際に結合エネルギーの算出(AI予測値)を行い、AIによる予測値と、DFT計算による正解値を比較することで、予測モデルの精度を評価しました(図2③)。
様々な手法で予測モデルを構築し、合計36個のモデルでの精度を比較した結果、最も高精度な予測モデルでは、僅かな平均誤差 (1.58 kcal/mol)での予測が可能でした。
- 研究成果2- 学習モデルの適応範囲の調査
適応範囲の調査では561種の超原子価ヨウ素の結合エネルギーをDFT計算によって算出し、AIでの予測値との比較によって精度の評価を行っています。この561種のDFT計算を1コア(1つのパソコンの1つの脳)で計算する場合に必要な時間は、4,272日、すなわち約12年間ですが、構築したAIによる予測では、561種すべての結合エネルギーを算出するのに、わずか2.9秒しか必要としませんでした。これは、従来のDFT計算よりも1.3億倍速いということになります。
以上のように、高性能なパソコンも高価なソフトウェアも専門知識を必要とせず、一般的に流通する個人のパソコン上で、分子の構造名を入力するだけで超原子価ヨウ素の結合エネルギーを算出する学習モデルの開発に成功しました。
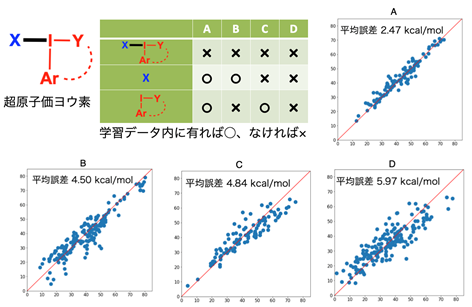 図 3 適応範囲の調査結果
図 3 適応範囲の調査結果
- 研究者のコメント(千葉大学大学院薬学研究院 中島誠也 助教)
- 研究プロジェクトについて
- 論文情報
・ 雑誌名:Scientific Reports
・ DOI: https://doi.org/10.1038/s41598-021-99369-8
- 用語解説
注2)DFT計算:密度汎関数理論(Density Functional Theory)。電子密度やエネルギーなどの分子や原子の物性を計算することが可能。
このプレスリリースには、メディア関係者向けの情報があります
メディアユーザーログイン既に登録済みの方はこちら
メディアユーザー登録を行うと、企業担当者の連絡先や、イベント・記者会見の情報など様々な特記情報を閲覧できます。※内容はプレスリリースにより異なります。
すべての画像
